The Ministry of Health, Labor and Welfare (MHLW) has been undergoing major restructuring, drastically altering the regulatory requirements and procedures for registering medical devices in Japan and Pharmaceuticals and Medical Devices Agency (PMDA) was established in April 2004 to create a more efficient and transparent registration review process, and subsequently new Pharmaceutical Affairs Law (PAL) went into effect on April 1, 2005.
In accordance with the enforcement of the revised PAL, application categories were changed so that they are now based on whether clinical data is required and whether approval standards exist. Low-risk medical devices for which certification standards have been established are now certified by a third party instead of being approved by MHLW. General medical devices are submitted to the PMDA
They have been actively incorporating approval review system that complies with specifications such as those of the International Organization for Standardization (ISO) and the International Electrotechnical Commission (IEC), as well as Summary Technical Documentation (STED) for applications base on the Global Harmonization Task Force (GHTF) formed by Japan, the US, EU, Australia and Canada.
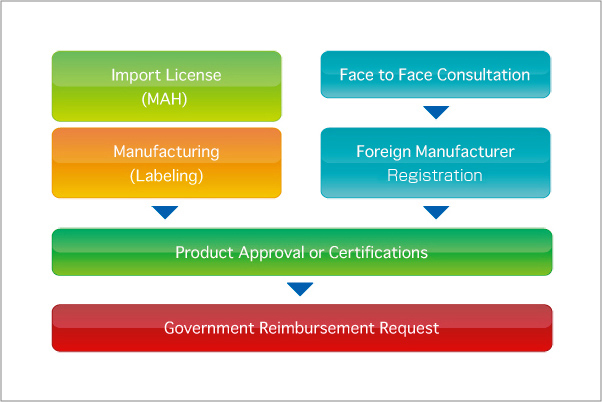
For registration of medical devices in Japan, there are 3 key elements, namely Manufacture, Product and Importer. Any manufacturer in foreign country who makes medical devices and wishes to export to Japan needs to obtain foreign manufactures accreditation from MHLW before product approval. Once this application is filed and accepted, product registration process can be proceeded to. The third key element is the import license holder in Japan as shown in the graphical Step Guides of Registration.
The new regulations has come into force since 25 November 2014,
and this amendments to Pharmaceutical Affairs Law(PAL)
includes expanded third-party certification for some Class ćV
devices, new regulatory requirements for certain stand-alone
medical software, simplification of medical device licensing,
and streamline PAL quality management system(QMS) requirements.
Details of major amendments are as follows:
üEExpansion of private third-party certification to include some highly
controlled Class ćV devices.
üEStand-alone regulatory requirement for software and programs
used to process or store medical
ü@data.
üEShifting manufacture licensing and accrediting system for foreign
manufacturing facilities to
ü@registration system.
üEStreamlining quality system inspections to focus on units of product
lines rather than individual.
üENewügRegenerative Productühcategory for product not easily classified
as either drugs or devices.
"Click each box for details"